Авторитетность издания

Добавить в закладки
Следующий номер на сайте


Разработка объединенной среды для анализа и поиска микрорнк
Аннотация:
Abstract:
| Авторы: Нечкин С. () - , Пальянов А. () - , Черемушкин Е. () - , Штокало Д. () - , Альберт П. () - , Лоренс ДЖ. () - | |
| Ключевые слова: анализ, поиск, биоинформатика, микрорнк |
|
| Keywords: analysis, search, , |
|
| Количество просмотров: 9846 |
Версия для печати Выпуск в формате PDF (8.40Мб) |
Авторами разработана программная среда для решения задач биоинформатики, связанных с молекулами, называемыми микроРНК. МикроРНК – это одноцепочечная молекула РНК длиной около 21–23 нуклеотидов, регулирующая производство белка. Эта молекула связывается с определенным участком мРНК, называемым сайтом связывания микроРНК, и блокирует его трансляцию. В настоящее время у человека экспериментально подтверждено наличие около 670 микроРНК. По разным оценкам, количество различных микроРНК человека варьируется в пределах от 1000 до 5000. К задачам анализа микроРНК относятся поиск и определение микроРНК, поиск сайтов посадки микроРНК, определение вторичной структуры и альтернативных вариантов вторичных структур комплексов микроРНК и мРНК, вторичных структур предшественников микроРНК. Эти молекулы открыты сравнительно недавно и вызвали огромный интерес у научного сообщества. Количество статей, алгоритмов и программ, посвященных им, стремительно растет. Негативным последствием столь бурного роста является несоответствие форматов ввода и вывода данных в различных программных системах. Исследователю-биологу приходится проводить полуручную обработку большого объема данных для получения научных результатов: отыскивать сайты микроРНК или определять вторичную структуру, а также объединять различные алгоритмы. Вместе с этим возникла необходимость в интеграции различных алгоритмов и программ работы с микроРНК в единую среду. Группой авторов совместно с институтом St.Laurent Institute (США) создана среда для интеграции наиболее популярных алгоритмов и на ее базе реализованы два собственных алгоритма.
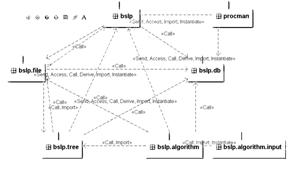
Описание программной системы. В рамках ведущегося проекта была реализована программная система для интергации различных алгоритмов работы с последовательностями микроРНК. Ввиду необходимости поддержки многопользовательского режима было решено разработать веб-приложение. Необходимые требования к программе – удобство в использовании, расширяемость, многофункциональность, возможность решения задач исследования микроРНК. Программный пакет содержит 40 классов на java и состоит из следующих основных блоков.
Рис. 1 1. Системные алгоритмы (запуск, контроль работы, остановка). 2. Работа с данными (загрузка, преобразование форматов, манипуляции с таблицами, экспорт). 3. Интерфейс (отображение результатов, таблиц, дерева файлов). 4. Последовательности действий – пайплайны (реализация соответствий). Диаграмма пакетов показана на рисунке 1. Опишем подробно концепцию менеджера процессов. Менеджер процессов – это управляющее процессами приложение, не зависящее от интерфейса. После того как пользователь выбирает параметры процесса и входные данные в среде, создается запись в таблице процессов в базе данных о том, что процесс подготовлен к запуску. Менеджер процессов сканирует таблицу процессов и, увидев эту запись, открывает новый поток для запуска и контроля процесса, меняет статус процесса на «выполняется». После удачного или неудачного завершения процесса изменяется его статус. Интерфейс программы представляет собой веб-приложение (рис. 2), содержащее структурированное по типу и алгоритму дерево файлов, основное окно, в котором отображаются исходные данные в виде таблицы или же результаты в виде таблицы или рисунка. В стадии реализации находится стратегия пайплайнов (конвейеров). Пайплайны представляют собой последовательности запускаемых алгоритмов, в которых вывод предыдущих шагов может использоваться как ввод следующих. Таким образом, изменив один из параметров в начале или середине цепочки алгоритмов, можно повторить весь процесс анализа и увидеть результат за один шаг.
Рис. 2 Описание алгоритмов. Алгоритмы, подключенные к среде, разделены на три группы. · Алгоритмы, разработанные в рамках пакета программ Vienna [1]: - RNADuplex считывает две последовательности РНК и вычисляет оптимальную и субоптимальные вторичные структуры для их гибридизаций (совпадений); - RNAFold предсказывает вторичную структуру с минимальной энергией и вероятности пар в РНК; - RNASubopt предоставляет полный субоптимальный фолдинг данной РНК. · Другие алгоритмы: - RNAhybrid. RNAhybrid [2] – программа для поиска гибридизации с минимальной свободной энергией для длинной и короткой РНК (выполняется в качестве доменной гибридизации, то есть короткие фрагменты позиционируются на лучший участок длинного фрагмента); - Teiresias – пакет программ для поиска комбинаций паттернов [3] (осуществляет поиск общих сигналов в выборке с учетом возможных разрывов в сигнале); - Blat – поиск гомологичных (схожих) участков в геноме для ДНК или РНК малой длины; · Алгоритмы, реализованные группой авторов: - RNAdraw позволяет осуществлять визуализацию молекул РНК с заданной вторичной структурой; - Functional Analysis определяет, насколько набор генов с найденными на них сайтами микроРНК соответствует определенному сигнальному пути из базы данных KEGG [4] или любой другой. Алгоритм был использован в проекте ExPlain [5]; - Enrichment позволяет анализировать, насколько гены данного сигнального пути обогащены сайтами микроРНК. Остановимся подробнее на алгоритме Functional Analysis. Исходными данными для него являются сигнальный путь S, содержащий набор генов G1,..,GN, и список генов с найденными на них сайтами микроРНК. Для каждой микроРНК Mi имеем Ni генов, на которых найдены сайты данной микроРНК: Gi1,…,GiNi. Далее для каждой Mi производится сравнение списков генов G1,..,GN и Gi1,…,GiNi. Вычисляется вероятность получения такого соответствия по случайным причинам (p-value). Таким образом, определяется, насколько данная микроРНК Mi регулирует гены данного сигнального пути S. В результате проделанной работы была реализована программная система, объединяющая различные алгоритмы работы с микроРНК. Система продолжает совершенствоваться и сегодня используется биологами. Список литературы 1. Vienna RNA secondary structure server Ivo L. Hofacker Nucleic Acids Research, 2003, Vol. 31, No. 13 3429-3431. 2. Marc Rehmsmeier *, Peter Steffen, Matthias Höchsmann, Robert Giegerich. Fast and effective prediction of microRNA/target duplexes /RNA, 10:1507-1517, 2004. 3. Rigoutsos, I. and A. Floratos, Combinatorial Pattern Discovery in Biological Sequences: the TEIRESIAS Algorithm. Bioinformatics, 14(1), January 1998. 4. Kanehisa M., Araki M., Goto S., Hattori M., Hirakawa M., Itoh M., Katayama T., Kawashima S., Okuda S., Tokimatsu T. and Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480-D484 (2008). 5. Kel A., Voss N., Jauregui R., Kel-Margoulis O. and Wingender E. Beyond microarrays: Find key transcription factors controlling signal transduction pathways/ BMC Bioinformatics, 7(Suppl. 2), S13 (2006). |

| Постоянный адрес статьи: http://swsys.ru/index.php?id=1651&like=1&page=article |
Версия для печати Выпуск в формате PDF (8.40Мб) |
| Статья опубликована в выпуске журнала № 4 за 2008 год. |
Возможно, Вас заинтересуют следующие статьи схожих тематик:
- Алгоритм анализа трафика в корпоративных компьютерных сетях на основе статистики экстремальных значений
- Основы структурно-лингвистического подхода в анализе нечетких временных рядов
- Препроцессорная обработка множеств прецедентов для построения решающих функций в задачах классификации
- Автоматизированная система поиска физических эффектов по запросу на естественном языке
- Кластеризация документов проектного репозитария на основе нейронной сети Кохонена
Назад, к списку статей