Journal influence
Bookmark
Next issue
Software suite for calculations of thermodynamic, spectroscopic and structural molecule properties
The article was published in issue no. № 1, 2011Abstract:The integrated program suite for calculations of structural, spectroscopic and thermodynamic individual substance properties is created. The vibration frequencies and thermodynamic properties are defined by means of developed algorithms for Lagrange's equation solution in curvilinear coordinates, and calculations of the statistical sum components for all freedom degrees. The suite is realized in the environment of object-oriented programming system Delphi.
Аннотация:Создан единый программный комплекс по расчету структурных, спектроскопических и термодинамических ха-рактеристик индивидуальных веществ. Частоты колебаний и термодинамические свойства определяются с помощью разработанных алгоритмов решения уравнения Лагранжа в криволинейных координатах и расчета составляющих статистической суммы для всех степеней свободы. Комплекс реализован в среде объектно-ориентированного про-граммирования Delphi.
| Authors: (turtsma@tversu.ru) - , Ph.D, (Yurij.Orlov@tversu.ru) - , Ph.D | |
| Keywords: spectral problem, thermodynamic properties, Lagrange's equation, molecule vibrations |
|
| Page views: 12266 |
Print version Full issue in PDF (5.09Mb) Download the cover in PDF (1.32Мб) |
Для оптимизации химических процессов и поиска новых технологий необходима информация о термодинамических свойствах веществ в широком диапазоне температур и давлений. К наиболее важным из них относятся энтальпия образования DfН°, свободная энергия Гиббса DfG°, энтропия S° и теплоемкость Cp° [1]. Информация, требуемая в необходимом объеме, может быть получена на основании эффективных расчетных методик, использующих реперные экспериментальные данные. Задача разработки таких методик и их реализации в программных комплексах одна из наиболее актуальных.
Расчет структурных характеристик начинается с определения локальных систем координат, центрированных на атомах. Геометрические параметры – положение атомов в декартовых координа- тах (
где q – вектор естественных координат, Внутренние координаты – это значения длин связей, валентных и двухгранных углов, естественные координаты q – их приращения. Пользователь может контролировать согласованность внутренних координат и при необходимости проводить корректировку. На заключительном этапе программа переносит начала всех систем координат в центр масс, дополняет набор базисных векторов до замкнутой системы и определяет единое многомерное пространство естественных координат. В построении учитываются все возможные естественные координаты, в том числе и линейно зависимые, то есть строится расширенная система естественных координат [2]. Это позволяет впоследствии выбрать те естественные координаты, для которых существует (или может быть подобран) набор силовых постоянных, однако существенно увеличивает число элементов матриц и решаемых уравнений. Программа способна вычислить структурные характеристики в системе СИ, в системе атомных единиц и с использованием спектроскопических масс водородов. Решение спектральных задач происходит после завершения построения геометрии соединения. Определение частот колебаний в гармоническом приближении осуществляется посредством решения уравнения Лагранжа в естественных (криволинейных) координатах. При этом матрица кинетической энергии вычисляется через матрицу кинематических коэффициентов [2, 3], в которую входят производные по времени от естественных координат (q). Элементами матрицы потенциальной энергии являются силовые постоянные. Набор силовых постоянных допускает определенный произвол в значениях и должен быть согласован с ранее выбранной совокупностью внутренних координат. Это делается либо вручную по подсказкам программы, либо путем отбора из табулированных данных силовых постоянных. Кроме того, для стандартных табулированных коэффициентов возможен автоматический ввод из базы силовых постоянных (дополнительно подгружаемого модуля). После задания силового поля производится расчет колебательного спектра. Порядок матриц кинетической и потенциальной энергий значительно превышает число независимых уравнений (3N-6, N – число атомов). Решение полученной системы уравнений заключается в одновременной диагонализации этих матриц. Матрица кинетической энергии приводится к единичной, а на диагонали матрицы потенциальной энергии получают значения квадратов частот. Однако количество элементов вектора собственных чисел превышает 3N-6, он содержит большое число малых величин, близких к значениям квадратов наименьших частот. Процедуры экспертного модуля, исходя из условий прямой задачи, отбирают требуемое число истинных квадратов частот и проводят предварительное отнесение по формам колебаний. Одновременно с этим комплекс TUR 7 вычисляет производные частот по силовым постоянным. Это позволяет определить долю потенциальной энергии колебаний, приходящуюся на каждую частоту, точно отнести найденные моды по видам колебаний и указать ядра, участвующие в колебательном движении с данной частотой. При вычислении термодинамических параметров по известному полному спектру часть предыдущих шагов можно опустить и вводить экспериментальные частоты вручную. Расчет термодинамических свойств соединений обычно требует знания потенциальных функций внутренних вращений V(j) и соответствующих им структурных характеристик. Химические связи, вокруг которых происходит вращение, выбираются в автоматическом режиме исходя из строения, за исключением вращения вокруг центрального атома, где нельзя указать связь вращения. В автоматическом режиме указывается только число симметрии волчка. При расчете вкладов внутренних вращений для каждого из них определяются атомы, составляющие остов и волчок, вычисляются моменты вращения остова, волчка и приведенный момент вращения (Ir). Для симметричных волчков приведенный момент соответствует Расчет S°(T), Cp(T), Н(Т)-Н(0), G(Т)-Н(0), большой статистической суммы и температурных зависимостей термодинамических свойств производится исходя из геометрического строения, спектра, потенциальных функций V(j) и приведенных моментов внутреннего вращения. Таким образом, процедуры этого модуля начинают выполняться только после завершения работы первых трех модулей. Найденные величины обычно представляются в виде таблиц. Одновременно с выводом значений свойств программа выдает все вклады в эти свойства: поступательный, вра- щательный, колебательный и вклад внутренних вращений. При выборе одной определенной температуры (обычно T=298 К) свойства представляются в виде единой большой таблицы. При вы- боре интервала и шага температур (например, T=298÷1500 К и DT=10 K) пользователю предоставляется выбор: рассчитать все свойства [S°(T), Cp(T), Н(Т)-Н(0), G(Т)-Н(0)] согласно введенному шагу или указать определенные из них (со значительным сокращением времени счета). Числа для каждого свойства даются в виде таблиц с указанием температур и вкладов. Отметим, что вклады рассчитываются с учетом нетемпературных поправок как для молекул, так и для радикалов (например, числа симметрии, поправки на оптические изомеры и мультиплетность). Значения теплоемкости в требуемом интервале температур представляются также графически. Зависимость Cp(T) аппроксимируется квадратичным, кубическим и обратно-квадратичным рядами: Сp=a+bT+cT2, (2) Сp=a+bT+cT2+dT3, (3) Сp=a+bT+cT2+dT–2. (4) Аналитическое представление (2–4) позволяет рассчитать Cp(T) для любой температуры заданного интервала и использовать соотношения Cp(T) в системах дифференциальных уравнений, описывающих химические процессы. Другая часть входных данных представляет V(j) и частоты колебаний [1]. Для многоатомных соединений полные и отнесенные по форме и симметрии колебательные спектры известны с удовлетворительной точностью (среднеквадратичная или средняя абсолютная погрешность менее 25 см-1) для относительно небольшой части молекул и единичных радикалов. Это связано с экспериментальными трудностями, вследствие чего отбор частот осуществляется при сопоставлении данных квантовохимических расчетов или модельного силового поля. Квантовохимический расчет колебательного спектра в ангармоническом приближении является очень ресурсоемкой задачей, поэтому с увеличением количества ядер и электронов приходится уменьшать степень учета электронной корреляции и число базисных функций, но при этом погрешности вычисленных частот также растут и в некоторых случаях превышают 100 см-1. Такая ошибка не позволяет охарактеризовать полученные на их основе свойства даже в качестве предварительных, процедуры из комплекса TUR 7 позволяют обойти и эту трудность. Использование в программе алгоритмов [2, 3] и локальных систем координат объясняется тем, что для них методические погрешности крайне малы, а систематическая погрешность в гармоническом приближении связана исключительно с погрешностью исходных данных (параметрами равновесного строения, потенциальными функциями и коэффициентами валентно-силового поля). В тех случаях, когда экспериментальные данные по равновесному строению отсутствуют, а квантовый расчет затруднен, геометрические параметры берутся как усредненные экспериментальные значения длин связей, валентных и торсионных углов родственных соединений. Многочисленные расчеты с TUR 7 показывают, что использование этого приема слабо сказывается на конечных результатах в сравнении с ошибками других вкладов. Кроме описанных модулей, предназначенных для решения отдельных, хотя и связанных задач, программа включает набор дополнительных модулей, позволяющих как получить всевозможные характеристики молекул, не связанные с основными расчетами, так и упростить работу с программой. На каждом этапе любые данные можно скопировать во внутренний буфер Windows с последующей вставкой в соответствующие программы, например, в документы Word или Excel. Графики копируются в форматах WMF, EMF или BMP. Все матрицы и таблицы (включая промежуточные) загружаются в буфер в форматах MAPLE, MatLab, а также в текстовом формате. Это позволяет интегрировать TUR 7 в другие программные продукты без значительных изменений и автоматизировать дальнейшие вычисления. Таким образом, средствами математического моделирования реальных процессов колебаний, происходящих в молекулярных системах, с использованием формализма криволинейных координат, метода вращений Якоби для приведения матриц к диагональному виду созданы эффективные алгоритмы, позволяющие находить решения громоздких избыточных систем уравнений Лагранжа (с числом уравнений в несколько сотен). Соединение в одном программном комплексе статистических методов и теории колебаний дало возможность находить термодинамические свойства индивидуальных веществ в любом температурном интервале. Алгоритмы, реализованные в программном комплексе TUR 7, вместе с его интерфейсом являются эффективным инструментом для проведения расчетных оценок важных физических величин. Литература 1. Годнев И.Н. Вычисление термодинамических функций по молекулярным данным. М.: Гостехтеориздат, 1956. 2. Gribov L.A., Orville-Thomas W.J. Theory and methods of calculation of molecular spectra. Chichester, NY: Jon Wiley and sons, 1989. 3. Грибов Л.А., Дементьев В.А. Методы и алгоритмы вычислений в теории колебательных спектров молекул. М.: Наука, 1981. 4. East A.L., Radom L. Ab initio statistical thermodynamical models for the computation of third-law entropies // J. Chem. Phys., 1997. V. 106. № 16, p. 6655. 5. Иориш В.С. Компьютерные методы расчета статистических сумм молекул и систематизации данных о термодинамических свойствах индивидуальных веществ: дисс… докт. хим. наук. М., 1995. |
 Авторами в среде Delphi создан программный комплекс TUR 7 по расчету термодинамических (DfН°, DfG°, S°, Cp°), спектроскопических (валентно-силовое поле, частоты колебаний) и структурных свойств молекул, представляющий собой совокупность взаимосвязанных модулей и БД. Из совокупности модулей в TUR 7 следует выделить четыре (рис. 1), объединяемых в различных сочетаниях в зависимости от поставленных целей. Первый служит для решения прямой спектральной задачи – расчета колебательного спектра по валентно-силовому полю на основании строения и атомарного состава соединения. Второй модуль предназначен для решения обратной спектральной задачи – определения валентно-силового поля (набор силовых постоянных) – исходя из равновесного строения и экспериментальных частот с известными отнесениями по симметрии и видам колебаний. Решение прямой и обратной спектральных задач производится в системе естественных координат и требует перехода от декартовых координат к естественным, поэтому в состав TUR 7 входит дополнительный модуль пересчета из одной системы координат в другую. С помощью третьего модуля находят структурные характеристики соединения: тензор инерции, главные моменты инерции, параметр асимметричности, связи, вокруг которых происходит внутреннее вращение, приведенный момент инерции, направляющие косинусы и т.д. Четвертый используется для определения S°(T), Cp°(T), Н(Т)-Н(0), G(Т)-Н(0).
Авторами в среде Delphi создан программный комплекс TUR 7 по расчету термодинамических (DfН°, DfG°, S°, Cp°), спектроскопических (валентно-силовое поле, частоты колебаний) и структурных свойств молекул, представляющий собой совокупность взаимосвязанных модулей и БД. Из совокупности модулей в TUR 7 следует выделить четыре (рис. 1), объединяемых в различных сочетаниях в зависимости от поставленных целей. Первый служит для решения прямой спектральной задачи – расчета колебательного спектра по валентно-силовому полю на основании строения и атомарного состава соединения. Второй модуль предназначен для решения обратной спектральной задачи – определения валентно-силового поля (набор силовых постоянных) – исходя из равновесного строения и экспериментальных частот с известными отнесениями по симметрии и видам колебаний. Решение прямой и обратной спектральных задач производится в системе естественных координат и требует перехода от декартовых координат к естественным, поэтому в состав TUR 7 входит дополнительный модуль пересчета из одной системы координат в другую. С помощью третьего модуля находят структурные характеристики соединения: тензор инерции, главные моменты инерции, параметр асимметричности, связи, вокруг которых происходит внутреннее вращение, приведенный момент инерции, направляющие косинусы и т.д. Четвертый используется для определения S°(T), Cp°(T), Н(Т)-Н(0), G(Т)-Н(0).
 ), длины связей, валентные и двухгранные углы – задаются с помощью соответствующих базисных векторов согласно [2, 3]. Это значительно упрощает расчеты, так как из рассмотрения исключаются поступательное и вращательное движения молекулы как целого. Векторы в локальных системах образуют два подмножества: направленные вдоль химических связей (
), длины связей, валентные и двухгранные углы – задаются с помощью соответствующих базисных векторов согласно [2, 3]. Это значительно упрощает расчеты, так как из рассмотрения исключаются поступательное и вращательное движения молекулы как целого. Векторы в локальных системах образуют два подмножества: направленные вдоль химических связей ( ) и направленные перпендикулярно связям (
) и направленные перпендикулярно связям (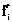 ) (рис. 2). Все входные данные автоматически переводятся программой во внутренние координаты, а затем в естественные:
) (рис. 2). Все входные данные автоматически переводятся программой во внутренние координаты, а затем в естественные: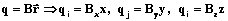 , (1)
, (1)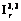 , для асимметричных
, для асимметричных 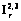 по классификации [4]. Свободное внутреннее вращение обрабатывается согласно [5]. Отметим, что программа допускает ввод параметров и для соединений с псевдовращениями. Все геометрические построения осуществляются в главных центральных осях, что позволяет при диагонализации соответствующих матриц и тензора инерции отделить внутреннее вращение от других степеней свободы.
по классификации [4]. Свободное внутреннее вращение обрабатывается согласно [5]. Отметим, что программа допускает ввод параметров и для соединений с псевдовращениями. Все геометрические построения осуществляются в главных центральных осях, что позволяет при диагонализации соответствующих матриц и тензора инерции отделить внутреннее вращение от других степеней свободы.| Permanent link: http://swsys.ru/index.php?id=2743&lang=en&page=article |
Print version Full issue in PDF (5.09Mb) Download the cover in PDF (1.32Мб) |
| The article was published in issue no. № 1, 2011 |
Back to the list of articles