Journal influence
Bookmark
Next issue
Structural analysis of rna sequences binding with HuR protein
The article was published in issue no. № 3, 2010Abstract:This work is devoted to development of computer methods for prediction of HuR binding sites. This RNA-binding protein HuR binds to specific regions of mRNA, which are usually located in 3’-UTR and are AU-reach (ARE). In this work we have implemented RNA secondary structure analysis and applied to analysis of experimental date provided by St. Laurent Institute.
Аннотация:Данная работа посвящена разработке компьютерных методов предсказания сайтов связывания белка HuR с определенными участками мРНК, которые, как правило, расположены в 3¢-нетранслируемом регионе и имеют повышенное содержание нуклеотидов A и U. Реализован учет вторичной структуры РНК и получен ряд новых результатов в ходе анализа экспериментальных данных, предоставленных институтом St. Laurent Institute (США).
| Authors: (evgeny.cheryomushkin@gmail.com) - , Ph.D, Cheremushkin E.S. (evgeny.cheryomushkin@gmail.com) - A.P. Ershov Institute of Informatics Systems (IIS), Siberian Branch of the Russian Federationn Academy of Sciences, Novel Computing Systems in Biology (Research Associate), Novosibirsk, Russia, Ph.D, (evgeny.cheryomushkin@gmail.com) - , (evgeny.cheryomushkin@gmail.com) - | |
| Keywords: mRNA expression regulation, algorithm, algorithm, RNA secondary structure, HuR, RNA-binding proteins |
|
| Page views: 11429 |
Print version Full issue in PDF (5.84Mb) Download the cover in PDF (1.43Мб) |
Белки семейства Hu (ELAVL) – HuB, HuC, HuD, HuR и ELAV, обладающие довольно высокой взаимной гомологией, связываются с ARE-элементами мРНК, расположенными в 3¢-нетранслируемом регионе, стабилизируя мРНК и увеличивая время ее жизни [1], то есть регулируют уровень экспрессии этой мРНК на уровне трансляции. Все белки семейства состоят из двух N-терминальных РНК-связывающих мотивов (RRM1 и RRM2), петлевого участка и С-терминального РНК-связывающего мотива RRM3. Механизм связывания представляет значительный интерес и активно изучается, однако полное понимание до сих пор не достигнуто. Например, in vitro HuR проявляет неизбирательную способность связываться с большинством ARE, а in vivo – только с определенными мРНК (на данный момент их известно примерно 34). Интерес к экспериментальному определению или компьютерному предсказанию остальных мишеней HuR по-прежнему велик. Ряд компьютерных методов уже был опробован исследователями [2], но не привел к решению проблемы в полной мере, возможно, по причине того, что поиск выполнялся только в пространстве последовательностей, не принимая во внимание вторичную структуру в районе ARE, которая может стать существенным фактором при взаимодействии белка с распознаваемым участком мРНК. Авторы предприняли попытку включить ее в рассмотрение при анализе данных. Описание алгоритма. В данной работе были использованы две выборки, предоставленные институтом St. Laurent Institute (Вашингтон, США). Первая выборка, TP=(sp1, …, spNp), позитивная, содержала последовательности РНК, показавшие связывание с белком HuR, а вторая – TN=(sn1, …, snNn) с отсутствием связывания. Длина каждой из последовательностей TP и TN варьируется от ста до нескольких тысяч нуклеотидов. На этих последовательностях был проведен поиск наиболее часто встречающихся вторичных структур и выбраны те, которые характеризуют каждую из выборок.
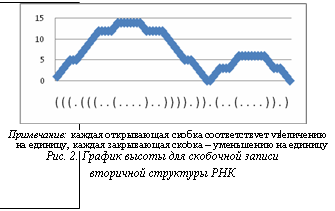
Алгоритм вычисления частоты структуры. Частота структуры Bij может быть вычислена путем сравнения ее с другими структурами Bkl, где k!=i. Сравнение структур производится путем вычисления редакторского расстояния между скобочными записями этих структур и сравнения его с заданным наперед порогом T (T=12). Но вычисление всех редакторских расстояний является очень трудоемкой задачей, поэтому было введено несколько оптимизаций, позволяющих исключить около 80 % сравнений, тем самым сделав алгоритм доступным для вычисления. С математической точки зрения эти оптимизации могут сделать расчет Fij неточным, но с биологической точки зрения учет этих оптимизаций дает наиболее достоверный результат. Перечислим оптимизации. 1. Выбор похожих по высоте структур. Для каждой структуры Bij построим график высоты (рис. 2) Hij(t), t=1, …, L(Bij), вычислив его следующим образом:
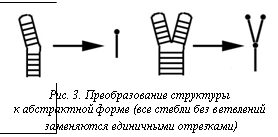
График показывает, что max(Hij) характеризует максимальную высоту структуры. Поэтому структуры, различающиеся по высоте более чем на DH=5, исключаются из сравнения. 2. Выбор структур, схожих по абстрактной форме. Чтобы уменьшить количество прямых сравнений структур, для каждой из них вычисляется абстрактная форма. В этой форме все ветви без ветвления заменяются единичными элементами (рис. 3). Сравнение абстрактных форм осуществляется значительно быстрее, поэтому выбирались структуры с одинаковой абстрактной формой. Пара структур, у которых абстрактная форма одинакова, но длины стеблей в АФ различаются более чем на D, исключается из сравнения. Используя описанные оптимизации и программу RNAdistance [4], для каждой структуры Bij получим искомую частоту Fij. В результате поиска структур с наибольшей частотой получены наборы структур для TP и TN. Каждой структуре поставлено в соответствие значение, определяющее количество последовательностей из TP и TN, содержащихся в данной структуре. Сначала были найдены структуры, которые встречаются в TP, но не в TN, а затем, наоборот, которые часто встречаются в TN, но не в TP. В результате полученные распределения структур были отсортированы по частоте встречаемости на последовательностях. Полученные распределения показаны на рисунке 4. В данной работе изучен структурный состав последовательностей РНК двух групп – связывающихся и не связывающихся с HuR. Эти последовательности отобраны экспериментальным путем в лаборатории института St. Laurent Institute. Был произведен поиск двухмерных структур РНК на этих последовательностях, а затем выбраны группы последовательностей. Несмотря на видимое различие в распределениях, необходимо заметить, что это различие необязательно является критерием для определения новых сайтов связывания с HuR. Некоторые эксперименты показывают, что это различие может быть вызвано отличием AU/GC состава последовательностей. В TP-выборке соотношение AU/GC=0,65, а в TF-выборке это соотношение равно 0,4. Пара AU образует двойную водородную связь, а пара GC – тройную. Поэтому структуры, образуемые последовательностями с разным составом AU/GC, будут различаться. Несмотря на это, по мнению авторов, исследование имеет ценность само по себе, без цели создать алгоритм распознавания HuR-сайтов. Литература 1. Peng S.S., Chen C.Y., Xu N., Shyu A.B. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO Journal. 1998. № 17, рр. 3461–3470. 2. Bolognani F., Contente-Cuomo T., Perrone-Bizzozero NI. Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res. 2010. № 38 (1), рр. 117–130. 3. Shapiro B., Zhang K. Comparing multiple RNA secondary structures using tree comparisons. Comput Appl Biosci. 1990. № 6, рр. 309–318. 4. Hochsmann M., Toller T., Giegerich R., Kurtz S. Local similarity in RNA secondary structures. Proceedings of the IEEE Bioinformatics Conference 2003 (CSB 2003). Standford University, USA. IEEE Computer Society, рр. 159–168. |

 Далее для каждой структуры Bij из B(si) была вычислена частота Fij встречаемости этой структуры на остальных последовательностях. То есть Fij – это процент последовательностей, на которых встретилась похожая структура. Структуры с наибольшей частотой являются структурами, характеризующими выборки, а также объектом поиска.
Далее для каждой структуры Bij из B(si) была вычислена частота Fij встречаемости этой структуры на остальных последовательностях. То есть Fij – это процент последовательностей, на которых встретилась похожая структура. Структуры с наибольшей частотой являются структурами, характеризующими выборки, а также объектом поиска.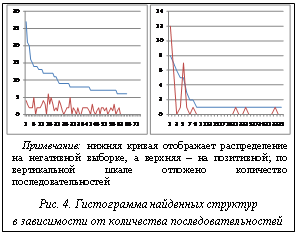

| Permanent link: http://swsys.ru/index.php?id=2584&lang=en&page=article |
Print version Full issue in PDF (5.84Mb) Download the cover in PDF (1.43Мб) |
| The article was published in issue no. № 3, 2010 |
Perhaps, you might be interested in the following articles of similar topics:
- Рекурсивный алгоритм точного расчета ранговых критериев проверки статистических гипотез
- Комплекс программ идентификации точечных дефектов листового стекла
- Эффективная программная реализация вейвлет-преобразования
- Способы реализации алгоритмов интегральных преобразований изображений по линиям
- Разработка алгоритмов функционирования математической модели дирижабельного радиолокационного комплекса обнаружения малозаметных воздушных целей
Back to the list of articles