Journal influence
Bookmark
Next issue
Library for transcription factor binding sites search
The article was published in issue no. № 4, 2011 [ pp. 110 – 113 ]Abstract:We developed library for searching transcription factor binding sites (specific fragments on DNA, where regulatory proteins binds. Those proteins are called transcription factors). Library consists of three search algorithms: direct search, permutation lookahead search and suffix arrays. Library is oriented on large scale information and allows performing search on genomic scale sequences with several thousand weight matrices.
Аннотация:Описывается разработанная авторами библиотека для поиска сайтов связывания с транскрипционными факторами (специфических фрагментов на ДНК, куда закрепляются регуляторные белки, называемые транскрипционными факторами). Она состоит из трех алгоритмов поиска: прямого поиска, поиска с использованием перестановок и за-глядывания вперед и суффиксных массивов. Библиотека ориентирована на большие объемы информации и позволя-ет производить поиск на последовательностях геномного масштаба с библиотекой из нескольких тысяч матриц.
| Authors: (evgeny.cheryomushkin@gmail.com) - , Cheremushkin E.S. (evgeny.cheryomushkin@gmail.com) - A.P. Ershov Institute of Informatics Systems (IIS), Siberian Branch of the Russian Federationn Academy of Sciences, Novel Computing Systems in Biology (Research Associate), Novosibirsk, Russia, Ph.D, (evgeny.cheryomushkin@gmail.com) - , (evgeny.cheryomushkin@gmail.com) - , Ph.D | |
| Keywords: software system, DNA, genome, metrics of test covering, transcription factor binding site, positional weight matrix |
|
| Page views: 15519 |
Print version Full issue in PDF (5.83Mb) Download the cover in PDF (1.28Мб) |
Регуляция генной экспрессии (производства РНК) на уровне транскрипции (считывания РНК) является очень сложным процессом, особенно в многоклеточных эукариотических организмах [1]. Каждый тип клетки или ткани на специфической стадии развития или под влиянием внеклеточных сигналов экспрессирует характеристический паттерн активированных транскрипционных факторов (ТФ). Внутри ядра клетки эти регулирующие транскрипцию белки связываются со специфическими сайтами связывания в регуляторных районах (промоторах и энхансерах) генов, способствуя их активации или репрессии. Компьютерные методы предсказания сайтов связывания с ТФ на ДНК очень важны для понимания молекулярных механизмов генной регуляции. Авторами была разработана библиотека, состоящая из трех различных алгоритмов предсказания сайтов, которые базируются на использовании весовых матриц (ВМ), собранных в БД TRANSFAC [2], и поэтому предоставляют возможность поиска огромного многообразия различных сайтов связывания с ТФ. Среди аналогичных программных продуктов можно выделить наиболее известные – SIGNAL SCAN [3], TESS [4]. Отличие разработанной авторами библиотеки состоит в том, что она позволяет искать сайты на последовательностях большого размера (вплоть до геномных масштабов), работает на обычном персональном компьютере и предоставляет только инструментарий для разработчика, не поддерживая графический интерфейс пользователя. Кроме того, в библиотеке, помимо простого поиска, реализованы ускоренные алгоритмы, использующие подход заглядывания вперед и перестановки, а также алгоритм, использующий суффиксные массивы для ускорения поиска. Рассматриваемые алгоритмы Весовые матрицы, описанные ранее в [5], представляют собой двухмерные массивы M=(mij), i=1, ..., 4, j=1, ..., N, составленные по выборке известных сайтов. На каждом фрагменте последовательности S длины N, S[k, …, k+N), может быть вычислен вес w весовой матрицы M с помощью следующей процедуры:
Далее производится нормировка веса Этот вес является характеристикой того, насколько данная последовательность схожа с последовательностями сайтов, используемыми для конструирования весовой матрицы, поэтому отражает вероятность, с которой данная последовательность может являться сайтом. Поиск потенциальных сайтов в последовательности S сводится к последовательному прикладыванию матрицы к каждой позиции k последовательности S и сравнению вычисленного веса wk с заданным наперед порогом c. В весовой матрице выделяется ядро – это наиболее консервативный фрагмент матрицы длиной Nc, зачастую равной 5. По ядру аналогично вычисляется вес ядра wck, который сравнивается с порогом ядра cc. Сайт считается распознанным в позиции k, если выполняются оба условия: wk³c и wck³cc. В авторской версии библиотеки реализованы обе возможности – поиск с ядром и без ядра. Разработанная библиотека содержит три алгоритма поиска сайтов, а также функциональность для чтения исходных данных (последовательностей и весовых матриц). Опишем эти алгоритмы. Алгоритм 1. Наивный поиск. Входная последовательность подается фрагментами, так как ее длина неизвестна и может быть очень большой. Если заданный фрагмент не является первым, поиск осуществляется с конца предыдущего фрагмента. Для этого после окончания поиска сохраняется конец последовательности длиной, равной максимальной длине матрицы: Поиск сайтов производится прямым вычислением веса на каждом фрагменте последовательности в режиме скользящего окна по формулам, приведенным выше. Если вес сайта выше порога, этот соответствующий объект заносится в массив распознанных сайтов. Алгоритм 2. Заглядывание вперед и пермутации. Построим индекс J по позициям в матрице таким образом: S(M, J(t))³S(M, J(t+k)) для k>0, где Таким образом, Sum_rest(M, t) – это сумма минимальных весов оставшихся позиций. Если последовательно вычислять частичные суммы весов Рассмотрим пример поиска с заглядыванием вперед и пермутациями. Пусть матрица М следующая:
Тогда вектор J будет таким:
На первой позиции стоит индекс 5, так как в 5-м столбце максимальный вес 12, больше чем в остальных столбцах, и т.д. Sum_rest будет следующим:
Sum_rest вычисляется с последнего элемента. Sum_rest(6)=5, так как в столбце с номером J(6)=2 максимум равен 5. Sum_rest(5)=5+6. И так далее складываются максимумы соответствующих столбцов. Пусть исходная последовательность следующая:
Порог C=40. Тогда последовательное вычисление частичных сумм выглядит таким образом:
Для вычисления W1 берем J(1)=5 – столбец матрицы и соответствующий нуклеотид S[5]=C. Соответствующий ему вес равен 12 и Wt+Sum_rest(t+1) дает 55, что выше порога С=40. Далее аналогичным образом вычисляются остальные веса. В позиции 4 суммарный вес оказывается ниже порога, что позволяет прекратить вычисления с ответом, что данная последовательность не является сайтом. Таким образом, используя этот подход, можно добиться значительного ускорения поиска сайтов. Но несмотря на это, скорость поиска все еще остается в линейной зависимости от длины последовательности. Следующий алгоритм позволяет сделать эту скорость логарифмической относительно длины последовательности. Алгоритм 3. Суффиксные массивы. Для этого алгоритма потребуется построить суффиксный массив по входной последовательности S. Суффиксный массив T содержит позиции i входной последовательности S, отсортированные в порядке возрастания суффиксов последовательности S. Так что, для некоторого L: S(T(i), L)>S(T(i+k), L) для k>0, где S(j, L) – фрагмент S, начиная с j длины L. Последовательность S считается законченной символом 0, меньшим любого другого символа из S. Строки сравниваются в лексикографическом порядке. Наиболее простой алгоритм построения суффиксного массива представляет собой построение суффиксного дерева. Опишем этот алгоритм. Шаг 1. Возьмем первую позицию и поместим ее в корневую вершину дерева Tr. Шаг 2. Для каждой следующей позиции i поместим ее в дерево Tr: а) если для некоторого L S(i, L) б) если для некоторого L S(i, L)>root, поместим i в правое поддерево. Выполним этот шаг рекурсивно для поддеревьев. Повторим шаг 2 для всех позиций i. Далее левосторонним обходом дерева получим желаемый массив. Рассмотрим пример построения суффиксного дерева для следующей последовательности: S=GCTCGATGC0. Индекс 1 попадает в корневую вершину. Подпоследовательность длины 1 следующего индекса 2 меньше корневой подпоследовательности C Таким образом, массив Т, полученный левосторонним обходом дерева, будет выглядеть так: T=(6, 9, 4, 2, 8, 5, 1, 3, 7). Далее сведем алгоритм поиска сайтов без ядра к алгоритму поиска с ядром. Длина ядра CL полагается равной 5. Найдем наиболее оптимальное ядро, в котором сумма максимальных весов максимальна: Вычислим порог ядра CW как минимальный порог, подходящий для ядра, за вычетом весов остальных нуклеотидов, то есть, если в остальных позициях вес будет максимальным, порог, определенный для ядра, найдем по следующей формуле:
Таким образом, поиск без ядра сводится к поиску с ядром. Теперь для каждой матрицы и каждого ядра построим набор допустимых последовательностей, для которых вес ядра больше порога. Алгоритм поиска заключается в последовательном переборе суффиксов. Берутся только те из них, которые входят в набор допустимых последовательностей, и по ним проводится проверка, подходит ли данная матрица по порогу к данной последовательности. За счет этого удается значительно ускорить поиск сайтов, но для последовательности необходимо предварительно построить суффиксный массив. Реализация Библиотека реализована на языке Java и включает в себя следующие классы. com.cogangs.bb.core.pwm TfMatrix – класс, реализующий функциональность весовой матрицы. Включает в себя методы получения весов, максимального и минимального веса по позиции, нормировки весов и другие вспомогательные функции. com.cogangs.bb.core.pwm.io LibMatrixReader – позволяет читать матрицы из входного файла в соответствующем формате. com.cogangs.bb.core.seq.io SAFileParser – читает и записывает суффиксный массив в бинарный файл соответствующего формата. Этот бинарный файл содержит как последовательность, так и суффиксный массив. Позволяет записывать последовательность по секциям. SuffixArrayBuilder – строит суффиксный массив по входной последовательности. com.cogangs.bb.core.tfbs NaiveSearcher – простой алгоритм поиска сайтов. Соответствует алгоритму наивного поиска. PALAlgorithms – вспомогательные функции, общие для алгоритмов заглядывания вперед, пермутаций и суффиксного алгоритма. PALSearcher – алгоритм с заглядыванием вперед и пермутациями. SASearcher – алгоритм поиска с суффиксными деревьями. TfbsResult – структура для хранения результирующих сайтов. В заключение следует отметить, что в данной работе были предложены три алгоритма для поиска потенциальных сайтов связывания с транскрипционными факторами с помощью весовых матриц. Алгоритмы ориентированы на большие объемы данных – последовательностей и матриц. Работа поддержана грантом COGANGS в рамках VII рамочной программы. Ф. Стейгмайер спланировал проект и управлял его разработкой. Литература 1. Gilbert SF. Developmental biology, 7th ed., Sunderland, Mass: Sinauer Associates, 2003. 2. Matys V. [et al.]. TRANSFAC: transcriptional regula- tion, from patterns to profiles Nucleic Acids Res. 2003. № 31, pp. 374–378. 3. Prestridge DS. SIGNAL SCAN 4.0: additional databases and sequence formats. Comput Appl Biosci. 1996. № 12(2). Apr., pp. 157–60. 4. Jonathan Schug, Using TESS to Predict Transcription Factor Binding Sites in DNA Sequence. Current Protocols in Bioinformatics, 2003. 5. Черемушкин Е.С. Программа для построения геномных профилей весовых матриц // Программные продукты и системы. 2010. № 4. |
 , где
, где
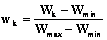 .
. .
.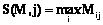 . То есть индекс J указывает на сортировку столбцов матрицы M по убыванию максимального элемента столбца. Далее вычислим остаточные суммы: Sum_rest(M, t)=
. То есть индекс J указывает на сортировку столбцов матрицы M по убыванию максимального элемента столбца. Далее вычислим остаточные суммы: Sum_rest(M, t)= где NM – длина матрицы M; J(j) – индекс, построенный ранее.
где NM – длина матрицы M; J(j) – индекс, построенный ранее. и если на текущем шаге t
и если на текущем шаге t 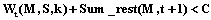 , то в дальнейшем, если даже в последующих позициях будут встречаться только максимальные веса, сумма весов все равно не превысит порога С (это ненормированный порог, вычисленный из равенства
, то в дальнейшем, если даже в последующих позициях будут встречаться только максимальные веса, сумма весов все равно не превысит порога С (это ненормированный порог, вычисленный из равенства 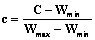 ). Таким образом, можно значительно сократить время поиска сайтов, отбрасывая на шагах вычисления веса сайты, заведомо меньшие порога. Индекс J называется пермутациями, а заглядывание вперед – это сравнение суммы с порогом на промежуточных шагах вычисления веса.
). Таким образом, можно значительно сократить время поиска сайтов, отбрасывая на шагах вычисления веса сайты, заведомо меньшие порога. Индекс J называется пермутациями, а заглядывание вперед – это сравнение суммы с порогом на промежуточных шагах вычисления веса.
 .
.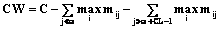 .
.| Permanent link: http://swsys.ru/index.php?id=2927&lang=en&page=article |
Print version Full issue in PDF (5.83Mb) Download the cover in PDF (1.28Мб) |
| The article was published in issue no. № 4, 2011 [ pp. 110 – 113 ] |
Perhaps, you might be interested in the following articles of similar topics:
- Программа для построения геномных профилей весовых матриц
- Comatch – поиск партнерских сайтов связывания с транскрипционными факторами
- Система поддержки многоатрибутивного принятия решений при управлении сложными системами
- Интеллектуальный подход к задаче информирования и краткосрочного прогнозирования временного ряда в онлайн-режиме
- Применение продукционной модели представления знаний для оценки соответствия уровня подготовки кандидата требованиям должности IT-отдела
Back to the list of articles